Departamento de Química e Bioquímica
Abstract
In this work we present a synthetic protocol that enabled the gram-scale preparation of key steroids intermediates designed to undergo a green fluorodecarboxylation. The produced intermediates can be used in the synthesis of important active pharmaceutical ingredients. This methodology was applied to a model drug, fluticasone propionate, a potent corticosteroid used in asthma treatment.
Keywords
Asthma
Fluticasone propionate
Fluorodecarboxylation
Bromofluoromethane
Introduction
Asthma is defined as a chronic inflammatory disorder of the airways characterized by paroxysmal or persistent symptoms such as dyspnea, chest tightness, wheezing, sputum production and cough, associated with variable airflow limitation and airway hyper-responsiveness to endogenous or exogenous stimuli.1 Asthma is a major cause of disability, health resource utilization and poor quality of life for those who are affected. It is the most common chronic disease among children and young adults, particularly because of its early onset (one out of four individuals in the general population develops asthma before the age of 40).2 Around 300 million people worldwide suffer from asthma, about 50% of whom live in developing countries,3 and approximately 250,000 people die prematurely each year. Almost all of these deaths are avoidable.4 It is estimated that the number of people with asthma will grow by more than 100 million by 2025. The exact cause of asthma is unknown but researchers think that it is caused by a combination of both genetic and environmental factors which contribute to its inception and evolution.5 Asthma costs money to governments.
In Europe, the total cost of asthma currently hovers at approximately €17.7 billion ($21.65 billion) per year.3 When the disease is controlled, there should be no more than the occasional recurrence of symptoms and severe attacks should be rare.6 Several classes of medication are available for the treatment of asthma, and often they must be taken concurrently to achieve asthma control.
Corticosteroid therapy, principally inhaled corticosteroid (ICS) therapy, is considered the most effective anti-inflammatory treatment.7 Fluticasone propionate (FP) is a potent ICS for the treatment of asthma. It is currently marketed in the United States as Flovent and in Europe as Flixotide. FP is available in both aerosolized metered dose inhaler (MDI) and dry powder devices. FP has been extensively studied in both children and adults. Efficacy of this corticosteroid has been documented across the entire spectrum of asthma severity, including corticosteroid-dependent disease.8
Subscribe to our e-Newsletters
Stay up to date with the latest news, articles, and events. Plus, get special offers
from American Pharmaceutical Review – all delivered right to your inbox! Sign up now!
Fluticasone propionate is synthesized in 16 steps, that includes an ozone depleting substance (ODS, bromofluoromethane) in the last step. Bromofluoromethane has an ODP of 0.73 (1 being the maximum) and therefore controlled by the Montreal Protocol. This Protocol was created under the Vienna Convention, which was a convention for the Protection of the Ozone Layer in Vienna in 1985, and where the nations agreed to “take appropriate measures to protect human health and the environment against adverse effects resulting or likely to result from human activities which modify or are likely to modify the Ozone Layer”. The Montreal Protocol was agreed on in 1987 and has since been amended five times in the light of developments in alternative technologies and substances and increasing scientific understanding of the status of ozone depletion and the processes involved. The Montreal Protocol has banned, or will eventually phase out, the production of all ODSs in all nations,9 for this reason, Hovione, a pharmaceutical company, has set up a program to find alternatives reagents to bromofluoromethane.
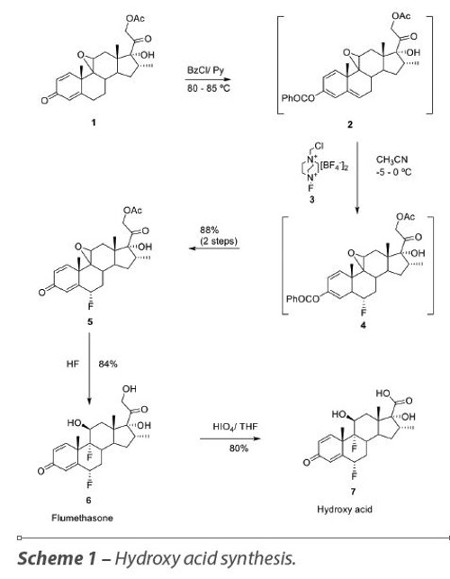
The processes for the synthesis of fluticasone propionate 12, starting from flumethasone 6, are known in the prior art. Flumethasone 6 is usually prepared starting from 17α,21-dihydroxy-9β,11b-epoxy-16α- methylpregna-1,4-diene-3,20-dione 21-acetate 1, which is a key synthetic intermediate for the synthesis of several glucocorticoids. Intermediate 1 reacts with benzoyl chloride (BzCl) to form the enolester 2, which reacts stereoselectively with an electrophilic fluorination agent (3) to introduce fluorine in the 6α-position, thus affording fluorinated intermediate 4. Deprotection of 4 at the C3 position leads to intermediate 5, which after reaction with HF and acetate hydrolysis gives flumethasone 6 with a purity higher than 95% by HPLC (% area, without purification). Oxidative cleavage of the hydroxymethyl group on C-17 of flumethasone 6 with periodic acid gives the hydroxy acid 7. The crude hydroxyl acid 7 obtained using this procedure has 96% purity by HPLC (% area) and after recrystallization from ethanol the purity increases to 99.2% purity (Scheme 1).10
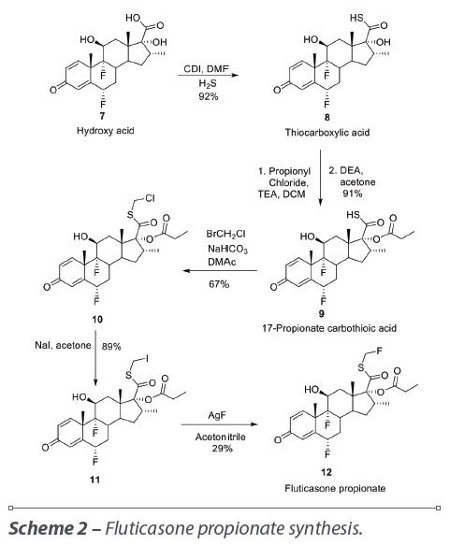
The hydroxy acid 7 is then activated with N,N’-carbonyldiimidazole (CDI) in dimethylformamide (DMF) and subsequent treatment with H2S gives the thiocarboxylic acid 8. The tertiary hydroxyl group of thiocarboxylic acid 8 is esterified with propionyl chloride and triethylamine (TEA) yielding the intermediate 9.11 Alkylation of 9 with bromochloromethane using NaHCO3 and dimethylacetamide (DMA) gives compound 10. Halogen exchange with NaI in acetone converts the chloromethyl ester 10 in iodomethyl ester 11, which in the presence of silver fluoride in acetonitrile forms fluticasone propionate 12 (Scheme 2).12
Several problems were encountered during this process such as difficulty removing traces of the chloromethyl ester 10 and the conversion of the chloromethyl ester 10 to iodomethyl ester 11, which produces impurities in significant amounts that are difficult to control/reduce to the limits of stringent pharmacopeial specification, even after recrystallization.
FP is usually prepared, as described in Israeli patent application IL 109656, directly from compound 9 using a halofluoromethane, such as bromofluoromethane. This process works very well and has the advantage of having a shorter synthesis and consequently a higher yield. However, it has the disadvantage of using a non-eco-friendly reagent (Scheme 3).
Our study was to find an intermediate that after fluorodecarboxylation would yield FP and also a suitable reagent to carry out the fluorodecarboxylation.
Results and Discussion
The study started with thioester 13. After a few attempts, the thioester 13 was obtained as the triethylamonium salt in quantitative yield with no need for purification (Scheme 4), by treatment of 17-propionate carbothioic acid 9 with triethylamine and bromoacetic acid in dichloromethane In the literature we can find a few methodologies to perform a fluorodecarboxylation. The Hunsdiecker13 and Kochi14 methodologies can be used to replace a CO2H function by a halide (Cl, Br, and I) but do not work with fluorine. The replacement of a carboxylic group with fluorine has previously been carried out with XeF2 15 or BrF3.16 When these procedures were tested with compound 13a complex mixture was obtained containing a small amount of the expected product (Scheme 5).
This result is perhaps predictable, as it is reported that a hydroxyl group can interfere with this reaction.17 Several reagents were used to protect the OH group at the 11 position such as: DHP (dihydropyran; with pTsOH in CH2Cl2 at 0ºC and ethoxyethene (with pTsOH in CH2Cl2 at 0ºC) the mixtures obtained in both cases were difficult to purify. The trifluoroacetate group is a suitable protecting group, under mildly acidic conditions and the cleavage of the trifluoroacetate can be accomplished selectively with ammonia without hydrolyzing the other esters in the molecule. Attempts to trifluoroacetylate substrate 9 with 1.2 equivalents of trifluoracetic anhydride did not afford the product but a complex mixture, and from the NMR spectrum we concluded that the thioester 13 was not present. Trifluoroacetic anhydride could have reacted with the carboxylic acid to form a mixed anhydride. Using 2.2 equivalents of the reagent did not improve the result and under the same conditions the 17-propionate carbothioic acid 9 reacted with trifluoroacetic anhydride as was indicated by a strong sulfurous smell and a complex mixture was obtained.
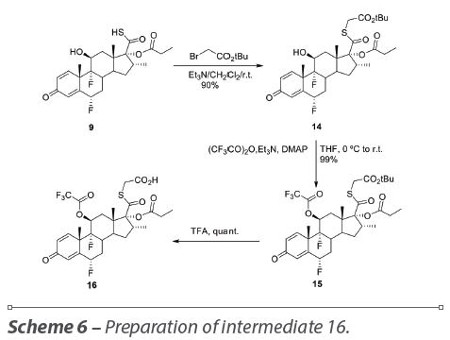
Realizing that the major problem in the formation of the trifluoroacetate was the presence of a free acid, the strategy was modified and tert-butyl bromoacetate was used to esterify the 17-propionate carbothioic acid 9 and compound 14 was obtained in quantitative yield. Protection, with trifluoroacetic anhydride, of the secondary hydroxyl group at the 11 position of compound 14, was first attempted without triethylamine as described in a Merck patent18 and product 15 was obtained in 56% yield. When we used triethylamine and DMAP (4-dimethylaminopyridine), the yield was 99%. Hydrolysis of the tert-butyl ester with TFA (trifluoroacetic acid) gave acid 16 in quantitative yield. These reactions were successfully scaled up to 8 g. All the reactions afforded a quantitative yield and only compound 15 was purified by flash chromatography, the other two products were obtained with high purity (Scheme 6).
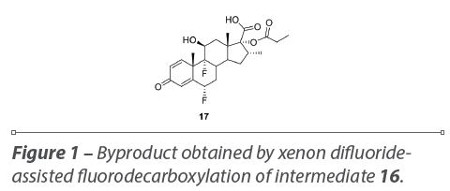
The fluorodecarboxylation reaction with xenon difluoride was extensively studied in order to optimize the yield of the reaction (Scheme 7). The amount of XeF2, the rate of addition, the temperature and reaction time were fully investigated. The best conditions were found to be the use of two equivalents of xenon difluoride, at -10 or 20 °C, for two days. At higher temperatures or prolonging the reaction time, extensive degradation occurred, producing 17-propionate hydroxy acid 17 as a byproduct formed by the cleavage of the thioester (Figure 1) with XeF2. This reaction was not purified and hydrolysis with ammonia 1M in methanol was performed in the crude product, affording FP and the yield of the two steps was 10%.19
Conclusions
A greener route for fluorodecarboxylation of steroids with pharmaceutical relevance has been developed as an alternative to the use of bromofluoromethane. The process relies on a XeF2-assisted, non-ozone depleting reaction. Fluticasone propionate was chosen as a model steroid and the obtained yield was lower than desired due to the low reactivity of starting materials and side reactions. Nevertheless, the protocol enables the efficient preparation of important new steroid intermediates in a gramscale, which can be used as a key material for the synthesis of other active pharmaceutical ingredients.
Experimental
1H NMR spectra were obtained at 400 MHz in CDCl3 or DMSO-d6 with chemical shift values (δ) in ppm downfield from tetramethylsilane. 13C NMR spectra were obtained at 100.61 MHz and 19F NMR spectra were obtained at 376.5 MHz. Assignments are supported by 2D correlation NMR studies. Some reactions were monitored by Waters High Performance Liquid Chromatographer (HPLC) model 600, equipped with auto sampler w717 plus and Photo Didode Array (PDA) detector W996. Medium pressure preparative column chromatography: Silica Gel Merck 60 H. Analytical TLC: Aluminium-backed Silica Gel Merck 60 F254. Reagents and solvents were purified and dried according to Purification of Laboratory Chemicals book.20
4.1. 2-(((6S,9R,10S,11S,13S,16R,17R)-6,9-difluoro-11-hydroxy-10,13, 16-trimethyl-3-oxo-17-(propionyloxy)-6,7,8,9,10,11,12,13,14,15,16,17-dodecahydro-3H-cyclopenta[a]phenanthrene-17-carbonyl)thio)acetic acid (13).
A solution of compound 9 (1 g, 2.1 mmol), triethylamine (0.440 mL, 3.15 mmol), bromoacetic acid (0.330 g, 2.31 mmol) in dichloromethane (10 mL) was stirred at room temperature overnight. Water was added (10 mL) and the mixture extracted with dichloromethane (3x10 mL), dried with anhydrous MgSO4, and concentrated to afford compound 13 (1.451 g, quantitative) as solid. 1H NMR (CDCl3, 400 MHz) δ (ppm): 7.22 (1H, d, J=10.1 Hz), 6.42 (1H, s), 6.37 (1H, dd, J=10.1, 1.6 Hz), 5.39 (1H, ddd, J=48.9, 10.3, 6.4 Hz), 4.38 (1H, d, J=9.08 Hz), 3.75 (1H, d, J=16.0 Hz), 3.65 (1H, d, J=16.0 Hz), 3.38-3.34 (1H, m), 3.12 (2H, dd, J=14.5, 7.2 Hz), 2.41-2.21 (5H, m), 2.02-1.98 (1H, m), 1.90-1.72 (2H, m), 1.53 (3H, s), 1.11 (3H, t, J=7.4 Hz), 1.11 (3H, s), 0.98 (3H, d, J=7.04 Hz). 13C NMR (CDCl3, 100 MHz) δ (ppm): 196.1, 185.8, 172.9, 172.6, 161.9, 161.8, 151.3, 129.9, 121.0, 120.9, 100.0, 98.2, 96.3, 86.5 (JCF=183 Hz), 71.7, 71.3, 60.4, 48.9, 48.2, 48.0, 45.7, 43.0, 36.2, 35.6, 34.1, 33.8, 33.6, 32.9, 32.8, 32.7, 32.6, 27.7, 23.0, 17.2, 16.1, 14.1, 9.1, 8.5.
4.2. (6S,9R,10S,11S,13S,16R,17R)-17-(((2-(tert-butoxy)-2-oxoethyl) thio)carbonyl)-6,9-difluoro-11-hydroxy-10,13,16-trimethyl-3-oxo-6,7,8,9,10,11,12,13,14,15,16,17-dodecahydro-3Hcyclopenta[a]phenanthren-17-yl propionate (14).
A solution of thiocarboxylic acid, compound 9 (5 g, 9.5 mmol), trimethylamine (2.2 mL, 14.2 mmol), tert-butyl bromoacetate (1.55 mL, 10.45 mmol) in dichloromethane (15 mL) was stirred at room temperature for 3h. Water was added (10 mL) and the mixture extracted with dichloromethane (3x 20 mL), dried with anhydrous MgSO4, and concentrated to afford compound 14 (5.6 g, quantitative) as a solid. Mp=102-103 ºC. 1H NMR (CDCl3, 400 MHz) δ (ppm): 7.14 (1H, dd, J=10.1, 1.2 Hz), 6.43 (1H, s), 6.37 (1H, d, I=10.1, J=1.8 Hz), 5.38 (1H, ddd, I=50.3, 13.2, 6.6 Hz), 4.42-4.39 (1H, m), 3.69 (1H, d, J=16.1 Hz), 3.60 (1H, d, J=16.1 Hz), 3.40-3.36 (1H, m), 2.69-2.68 (1H, m), 2.41-2.22 (6H, m), 1.97-1.74 (4H, m), 1.52 (3H, s), 1.48 (9H, s), 1.35-1.29 (1H, m), 1.15-1.10 (6H, m), 0.98 (3H, d, J=7.2 Hz). 13C NMR (CDCl3, 100 MHz) δ (ppm): 195.3, 185.5, 172.7, 167.9, 161.2, 150.5, 130.2, 121.2, 121.1, 99.4, 97.7, 96.1, 86.5 (JCF=183 Hz), 82.3, 72.0, 71.6, 48.6, 46.1, 42.9, 36.3, 36.2, 34.1, 33.7, 33.5, 32.8, 27.9, 27.6, 23.0, 17.2, 16.2, 9.0. FT-IR (KBr): 3457, 1745, 1670, 1631 cm-1. 4.3. (6S,9R,10S,11S,13S,16R,17R)-17-(((2-(tert-butoxy)-2-oxoethyl)thio) carbonyl)-6,9-difluoro-10,13,16-trimethyl-3-oxo-11-(2,2,2-trifluoroacetoxy)- 6,7,8,9,10,11,12,13,14,15,16,17-dodecahydro-3H-cyclopenta[a]phenanthren-17-yl propionate (15).
A solution of compound 14 (5.53 g, 9.5 mmol), triethylamine (3.29 mL, 23.7 mmol), trifluoroacetic anhydride (TFA) (3.29 mL, 23.7 mmol) and a catalytic amount of DMAP in THF (20 mL) was stirred at room temperature overnight. Water was added (15 mL) and the mixture extracted with ethyl acetate (3x20 mL), dried with anhydrous MgSO4, and concentrated. Purification byflash column chromatography (1:9 AcOEt/hexane – 4:6 AcOEt/hexane) afforded compound 15 (6.4 g, quantitative) as a solid. Mp=95-96 ºC. 1H NMR (CDCl3, 400 MHz) δ (ppm): 6.66 (1H, d, J=10.1 Hz), 6.46 (1H, s), 6.41 (1H, d, J=10.2 Hz), 5.58-5.56 (1H, m), 5.35 (1H, ddd, J=48.8, 11.5, 6.3 Hz), 3.63 (2H, s), 3.41-3.37 (1H, m), 2.47-2.15 (7H, m), 1.91-1.65 (3H, m), 1.43 (9H, s), 1.37 (3H, s), 1.13 (3H, t, J=7.5 Hz), 1.02-0.99 (6H, m). 13C NMR (CDCl3, 100 MHz) δ (ppm): 195.1, 184.5, 172.4, 167.4, 159.3, 159.2, 155.6, 155.2, 147.5, 131.4, 121.7, 121.6, 98.1, 96.3, 95.3, 85.9 (JCF=184 Hz), 82.3, 75.6, 75.2, 60.3, 48.0, 47.1, 46.8, 42.4, 36.2, 33.9, 33.5, 33.3, 33.2, 33.1, 33.0, 32.8, 32.4, 27.7, 27.5, 22.46, 22.42, 17.1, 15.7, 9.0. FT-IR (KBr): 1789, 1747, 1675, 1639 cm-1.
4.4. 2-(((6S,9R,10S,11S,13S,16R,17R)-6,9-difluoro-10,13,16-trimethyl-3-oxo-17-(propionyloxy)-11-(2,2,2-trifluoroacetoxy)-6,7,8,9,10,11,12,13,14,15,16,-17-dodecahydro-3H-cyclopenta[a]phenanthrene-17-carbonyl)thio)acetic acid (16).
A solution of compound 15 (6.4 g, 9.4 mmol) in TFA (15 mL) was stirred at room temperature for 2h. Evaporation of TFA afforded compound 16 as a solid (quantitative). Mp=246-246 ºC. 1H NMR (CDCl3, 400 MHz) δ (ppm): 9.73 (1H, broad s), 6.81 (1H, d, J=10.0 Hz), 6.56-6.51 (2H, m), 5.58-5.57 (1H, m), 5.38 (1H, ddd, J=48.3, 10.5, 6.2 Hz), 3.80 (1H, d, J=16.6 Hz), 3.74 (1H, d, J=16.6 Hz), 3.40-3.36 (1H, m), 2.47-2.30 (6H, m), 2.15-2.10 (1H, m), 1.93-1.78 (2H, m), 1.38 (3H, s), 1.13 (3H, t, J=7.5 Hz), 1.01 (3H, d, J=7.1 Hz), 0.97 (3H, s). 13C NMR (CDCl3, 100 MHz) δ (ppm) 195.0, 184.4, 173.5, 173.2, 173.0, 162.1, 161.9, 159.7, 159.2, 158.8, 156.1, 155.6, 155.2, 154.8, 149.9, 130.7, 121.2, 121.0, 98.2, 96.5, 95.5, 85.9 (JCF=185 Hz), 82.3, 75.5, 75.1, 61.2, 48.0, 47.6, 47.5, 47.3, 42.4, 36.3, 33.8, 33.5, 33.3, 33.2, 33.1, 33.0, 32.9, 32.4, 31.2, 30.4, 27.6, 22.1, 22.0, 21.0, 17.0, 15.7, 8.9. FT-IR (KBr): 3218, 1789, 1725, 1671, 1637 cm-1.
4.5. Fluticasone propionate (12).
To compound 16 (0.5 g, 0.80 mmol) in dichloromethane (40 mL) at -10 ºC it was added XeF2 (0.260g, 1.6 mmol) and the solution was stirred at -10 ºC for 2 days. 5% NaHCO3 aqueous solution was added (40 mL) and the mixture extracted with dichloromethane (3x 30 mL), dried with anhydrous MgSO4, and concentrated to afford a crude mixture containing compound 18 (0.390 g) as a solid foam which was not purified. This crude mixture in MeOH (2 mL) was treated with 1M NH3 solution in MeOH (0.783 mL) at 0 ºC. After 5 min, the volatiles were evaporated and flash column chromatography of the residue (1:9 AcOEt/hexane – 5:5 AcOEt/hexane) afforded compound of formula 12 (0.041 g, 10%, 2 steps) as a white solid. 1H-NMR (CDCl3), 400 MHz: δ 7.11 (1H, d, J=10 Hz), 6.44 (1H, s), 6.38 (1H, d, J=10 Hz), 5.93 (1H, dd, J=33.6 Hz, J=9.4 Hz), 5.80 (1H, dd, J=33.6 Hz, J=9.3 Hz), 5.38 (1H, ddd, J=49.4, J=11.4, J=6.4 Hz), 4.43-4.41 (1H, m), 3.41-3.38 (1H, m), 2.40-2.26 (6H, m), 1.92-1.73 (4H, m), 1.52 (3H, s), 1.37-1.31 (1H, m), 1.13 (3H, t, J=7.5 Hz), 1.09 (3H, s), 0.99 (3H, d, J=7.2 Hz). 13C NMR (CDCl3), 100 MHz: δ 193.0, 185.5, 172.9, 161.2, 161.1, 150.3, 130.3, 121.2, 121.1, 99.5, 97.8, 96.2, 86.4 (JCF=183 Hz), 80.8 (JCF=215 Hz), 72.0, 71.6, 48.5 48.0 47.8, 43.0, 36.5, 36.2, 34.0, 33.7, 33.5, 32.8, 32.7, 32.6, 32.5, 27.5, 23.0, 23.0, 17.1, 16.4, 9.0.
Acknowledgments
We thank Hovione for their financial support. We thank the Fundação para a Ciência e Tecnologia and MostMicro Research Unit (financially supported by LISBOA-01-0145-FEDER-007660 funded by FEDER funds through COMPETE2020 (POCI) and by national funds through FCT, and FCT PTDC/BIA-MIC/4188/2014). The NMR data was acquired at CERMAX, ITQBNOVA, Oeiras, Portugal with equipment funded by FCT, project AAC 01/ SAICT/2016.
References and notes
- Lougheed, M. D.; Lemiere, C.; Ducharme, F. M.; Licskai, C.; Dell, S. D.; Rowe, B. H.; FitzGerald, M.; Leigh, R.; Watson, W.; Boulet, L.-P., Canadia throracic society 2012 guideline update: diagnosis and management of astma in preschoolers, children and adults: executive summary. Can. Respir. J. Nov./Dec. 2012, 19 (6).
- To, T.; Wang, C.; Guan, J.; McLimont, S.; Gershon, A. S., Am. J. Respir. Crit. Care Med. 2010, 181, 337-343.
- Braman, S. S., The Global Burden of Asthma. Chest 2006, 130, 4S-12S.
- Spiro, S. G.; Silvestri, G. A.; Agustí, A., Clinical Respiratory Medicine. 4th ed.; Elsevier Health Sciences: 2012.
- Busse, W. W.; Lemanske, R. F. J. N., Engl. J. Med. 2001, 344 (5), 350-362.
- Holohan, J.; Manning, P.; Nolan, D., Asthma Control in General Practice. Adapted from the GINA Global Strategy for Asthma Management and Prevention. 2nd ed.; ICGP: 2013.
- Nelson, H. S., Advair: combination treatment with fluticasone propionate/salmeterol in the treatment of asthma. J. Allergy Clin. Immunol. 2001 Feb, 107 (2), 398-416.
- Staresinic, A. G.; Sorkness, C. A., Fluticasone propionate: a potent inhaled corticosteroid for the treatment of asthma. Expert Opin. Pharmacother 2000, Supl 1(6), 1227-1244.
- MOD Corporate Environmental. JSP 418. Protection Manual. Leaflet 7. February 2011. Ozone Depleting Substances.
- Villax, I.; Mendes, Z. Preparation of flumethasone and its 17-carboxyl androsten analogue. WO Pat. Appl. 2002100878 A1, June 12, 2001.
- (a) Phillipps, G. H.; Bain, B. M.; Steeples, I. P.; Williamson, C. Androstane carbothioates. US Patent 4,335,121, February 13, 1981; (b) Sobral, L.; Martin, D.; Heggie, W.; Leitao, E. Process for the Esterification of a Carbothioic Acid. US Patent 20070287846 A1, October 19, 2007.
- Leitão, E. P. T.; Heggie, W. Methods and compounds for the preparation of monofluoromethylated biologically active organic compounds. WO Pat. Appl. 2011151625 A1, June 1, 2010.
- Hunsdiecker, H.; Hunsdiecker, C. Method of manufacturing organic chlorine and bromine derivatives. US Patent 2,176,181, October 17, 1939.
- Kochi, J. K., J. Am. Chem. Soc. 1965, 87 (11).
- Timothy, B. P.; Kamalesh, K. J.; David, H. W., Can. J. Org. Chem. 1983, 48, 4158-4159.
- Halpern, D. F.; Robin, M. L. Method for fluorodecarboxylation. 1990.
- Patrick, T. B.; Johri, K. K.; White, D. H.; Bertrand, W. S.; Mokhtar, R.; Kilbourn, M. R.; Welch, M., J. Can. J. Chem. 1986, 64, 138-141.
- Ali, A.; Greenlee, M. L.; Mcvean, C. A.; Meissner, R. S.; Taylor, G. E. D homoandrosta-17-yl-carbamate derivatives as selective glucocorticoid receptor ligands. WO Pat. Appl. 2006124710 A1, May 18, 2005.
- Leitão, E. P. T.; Maycock, C.; Ventura, M. R. Method for the production of fluoromethyl – esters of androstan- 17 - beta - carboxylic acids. . WO Pat. Appl. 2012160338 A1, May 26, 2011.
- Armarego, W. L. F.; Chai, C. L. L., Purification of Laboratory Chemicals. 5th ed.; Elsevier: 2003.